| Home -> Miscellaneous Books -> Experimental Organic Chemistry -> Chapter I. - Laboratory Methods | |||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||
|
Chapter I
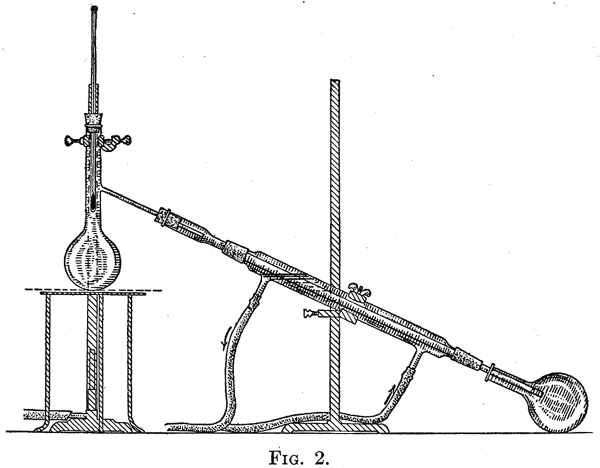
Laboratory Methods 1. General Directions to the Student. - Before beginning an experiment read through to the end the directions which are to be followed. Many mistakes which involve additional work can be prevented by understanding beforehand just what is to be done. The import of the experiment should be clear, and the chemical reactions involved at each step should be understood before the work is started. References are given in each experiment to the section in the author's text-book "The Principles of Organic Chemistry" in which the chemical reactions involved are discussed. These references are given in bold-face type thus, (Section 359). References to paragraphs in this book are indicated thus, §64, page 42. Keep a clear and concise record of the laboratory work. The notes should be written as soon as the experiment has been performed, and care should be taken to have the original record, made during the course of the experiment, of such a character that it serves as the permanent record of the work. Notes should not be taken on loose pieces of paper and afterward written out in the notebook; they should be written carefully in good English, and should state briefly what was done and what was observed. It is necessary for the student to recognize what the experiment is to teach - why he was asked to do it. If the work consists in the preparation of some compound the details for which are given in the laboratory guide, it is not advisable to take time to copy these details in the notebook. References to the pages in the book where the preparation is described should be given, and a statement made of the amounts of the substances used. If any unexpected difficulties arose, or if any improvement in the way of carrying out the preparation was used, these facts should be noted. Write equations for all reactions taking place in the experiment, and record the yield of the compound obtained. The substance should be put in a clean, dry, glasss-toppered bottle of appropriate size, and be labeled. The student's name, the name, weight, and the boiling-point or melting-point of the substance should be recorded on the label. The boiling-point or melting-point should be that observed by the student for the sample itself, and not the points recorded in the book. The student should use reasonable care in his manipulations. He should endeavor to get as large a yield as possible of the product sought, but should use judgment as to whether it is advisable to spend a large amount of time to increase by a small amount the yield of the product. The processes should not be carried out in the manner used with a quantitative analysis - a few drops may be lost here and there if they form but a very small portion of the total amount formed, and their recovery entails the expenditure of much extra time. It is not meant by this that the student be careless; he should develop judgment as to the relative value of a slightly higher yield of product and the time required to obtain it. 2. Calculation of Yield. - The student should calculate in each preparation the percentage yield obtained from the chemical equation for the reaction can be calculated the so-called theoretical yield. The percentage of this obtained is called the percentage yield. The latter is never equal to 100 per cent for many reasons. It is often advisable to use an excess over the theoretical amount of one of the substances used in the preparation. The student should, before calculating the percentage yield obtained, determine whether an excess of one reagent has been employed. When one substance used in a preparation is much more expensive than the rest, it is customary to take the substances in such amounts that the largest yield possible calculated from the more expensive substance is obtained. For example, preparations involving the use of iodine are so carried out that the largest amount of the halogen possible is obtained in the substance prepared. In this case the test of the skill with which the preparation is carried out is determined by this fact; the percentage yield should be calculated, accordingly, from the weight of iodine used. 3. Integrity in Laboratory Work. - The student should record in his notebook his own observations only, and the results he has obtained himself, unless there is a definite statement to the contrary. If a student has carried out an experiment along with another student a statement to this effect should be put into the notes. 4. Cautions in Regard to Laboratory Work. - A student uses in laboratory work in organic chemistry inflammable liquids and substances like sodium and phosphorus which have to be handled with great care. As a result fires may happen. The laboratory should be provided with buckets of sand and a fire-extinguisher. A heavy woolen blanket should be near at hand to be used in case the clothing catches fire. Inflammable liquids such as ether, alcohol, and benzene should not be poured into the jars provided for acids. Only cold solutions should be extracted with ether, and the process should be carried out at least twelve feet from a flame. When carrying out a reaction in a test-tube, care should be taken to hold the tube in such a position that if the contents are violently thrown out, they will not come in contact with the experimenter or any one in the neighborhood. If the odor of a substance in the tube is to be noted, do not look down into the tube. If this is done and a violent reaction takes place suddenly, the material in the tube may be thrown into the eye. Crystallization 5. When an organic compound has been prepared it must be purified from the by-products which are formed at the same time. In the case of solid substances crystallization is ordinarily used for this purpose, although with certain compounds purification can be more readily effected by sublimation or distillation, processes which are described below. Choice of Solvent - The separation of two substances by means of crystallization is based on the fact that they are present in the mixture to be separated into its constituents in different amounts, or on the fact that the two substances possess different solubilities in the liquid used as a solvent. When it is desired to purify a substance by crystallization a solvent should be selected, if possible, in which the impurity is readily soluble, and in which the substance sought is more or less difficultly soluble. Purification is effected most easily when the substance to be purified is appreciably soluble in the hot solvent, and much less soluble in it when cold. If the two conditions stated above can be combined - and this is possible in many cases - purification is readily accomplished. The solvents most commonly used in crystallization are water, alcohol, ether, benzene, petroleum ether, ligroin, carbon bisulphide, chloroform, acetone, and glacial acetic acid. In certain cases hydrochloric acid, carbon tetrachloride, ethyl acetate, toluene, and nitrobenzene have been found of particular value as solvents. In order to crystallize a compound the solubility of which is not known, preliminary tests should be made with the solvents enumerated above; about 0.1 gram or less of the substance should be used in each test. The solid is placed in a small test-tube, and the solvent is added a drop at a time and the tube is shaken. After the addition of about 1 cc. of the liquid, if the substance has not dissolved, the tube should be heated until the liquid boils. If the substance does not dissolve, more liquid should be added in small quantities until solution occurs. If a very large amount of the liquid is required for solution, or the substance proves insoluble, another solvent must be used. When solution takes place the tube is cooled by running water. If the substance separates, it is redissolved by heating, and the contents set aside to cool slowly, when crystals will probably form. If the substance does not separate to a considerable degree when the hot solution is cooled, similar tests should be made with other liquids. If none of the solvents can be used in this way, either the substance must be obtained by spontaneous evaporation, or a mixture of liquids must be used - a method described below. If the compound is to be crystallized by spontaneous evaporation, cold saturated solutions, prepared by dissolving about 0.1 gram or less of the substance in a number of solvents, are poured onto watch-glasses and left to evaporate slowly. 6. Some substances form solutions from which the first crystals separate with difficulty. In such cases the solution is "seeded" by adding a trace of the solid substance; a piece the size of the head of a small pin is sufficient. Crystallization of such substances can often be brought about by scratching with a glass rod the side of the vessel containing the solution; the rough surface so formed assists materially in the formation of the first crystal, after which crystallization proceeds readily. The liquid finally selected for the solvent should be one which yields well-formed crystals, and does not evaporate too slowly. 7. Use of Freezing Mixtures in Crystallization. - It often happens that substances which do not separate from their hot solutions when the latter are cooled with water, crystallize out well when the solutions are allowed to stand for some time in a freezing mixture. For this purpose, a mixture consisting of equal weights of sodium chloride and finely divided ice or snow, is commonly used; with snow a temperature of -17° is obtained. A mixture of equal weights of crystallized calcium chloride and snow gives the temperature -48°. A convenient freezing mixture is made by covering finely divided ice with commercial concentrated hydrochloric acid. 8. Preparation of Crystals. - When a satisfactory solvent has been selected, the material to be crystallized is placed in a beaker and covered with the liquid. The mixture is heated to boiling over a free flame or on a steam-bath if the solvent used is inflammable. It is essential to avoid the presence of a free flame when alcohol, benzene, ether, or petroleum ether are used as solvents. The beaker is covered with a watch-glass, and the solvent is added in small portions at a time until the substance to be crystallized has passed into solution. It may happen that a small amount of a difficultly soluble impurity is present; in this case it is not advisable to add enough solvent to dissolve the impurity. When the substance to be crystallized has been dissolved, the solution is filtered while hot through a fluted filter-paper into a beaker. Crystallizing dishes should not be used. If the substance crystallizes out during the filtration, either a hot-water funnel can be used, or enough of the solvent can be added to prevent crystallization. In the latter case, and whenever an excess of solvent has been used, it is advisable to concentrate the solution to crystallization after filtration. 9. The solution is evaporated to crystallization by boiling it gently. Tests are made from time to time to determine whether crystals will form when the solution cools. This can be readily done by placing a glass rod in the hot solution and then withdrawing it; if crystals appear when the drop of the liquid which adheres to the rod cools, the solution should be set aside and covered with a watch-glass or filter-paper. If crystals are not formed, the evaporation should be carried further. A hot-water funnel is at times very useful if crystals form during the filtration. It consists of a funnel surrounded by a metal jacket in which is placed water that can be heated to its boiling-point by means of a Bunsen burner. When inflammable liquids are used as solvents, the water should be heated and the burner extinguished before filtration. Disregard of this precaution has frequently led to fires. 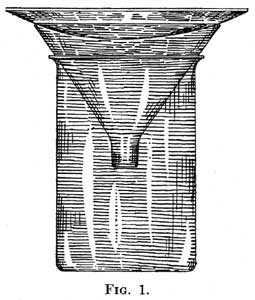 10. It is advisable to cut off the stems of the funnels to be used in the preparation of organic compounds. This eliminates the clogging of the funnel as the result of crystallization of solids in the stem. It also makes it unnecessary, in most cases, to use filter-stands as the funnel can be supported by the beaker which is to hold the filtrate; if the beaker is too large for this, the funnel can be supported on a clay triangle placed on the beaker. The arrangement represented in Fig. 1 is especially convenient for filtering solutions which deposit crystals on cooling slightly. During filtration the beaker is heated on the steam-bath or over a flame; the vapor which rises heats the funnel. The latter should be covered with a watch-glass to prevent loss of heat during filtration, from the liquid that it contains. 10. It is advisable to cut off the stems of the funnels to be used in the preparation of organic compounds. This eliminates the clogging of the funnel as the result of crystallization of solids in the stem. It also makes it unnecessary, in most cases, to use filter-stands as the funnel can be supported by the beaker which is to hold the filtrate; if the beaker is too large for this, the funnel can be supported on a clay triangle placed on the beaker. The arrangement represented in Fig. 1 is especially convenient for filtering solutions which deposit crystals on cooling slightly. During filtration the beaker is heated on the steam-bath or over a flame; the vapor which rises heats the funnel. The latter should be covered with a watch-glass to prevent loss of heat during filtration, from the liquid that it contains. |
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
16. Preparation of Corks. - Before being used corks should be softened. This can be done by means of a press, which is made for this purpose, or the cork can be rolled on the desk while it is being pressed firmly by means of a block of wood. It is, in most cases, not advisable to use rubber stoppers as they may be attacked by the vapor of the liquid during distillation. Sharp cork borers should be used to make the holes of such a size that the tubes to pass through fit snugly. In boring corks it is advisable first to push the borer with a rotary motion half way through the cork, taking care that the hole is bored through the center of the cork; the borer is then removed and a hole made from the center of the other end of the cork to meet that first made. By proceeding in this way the edges of the holes on the two sides of the cork will be clean cut, and thus make a tight joint with the tube to be passed through the hole; and the latter will run evenly through the axis of the cork. 17. Position of the Thermometer. - The bulb of the thermometer should be so placed that it is about 1 inch below the side arm of the distilling flask. If the liquid boils at such a point that the end of the thread of mercury is hidden by the cork during the boiling, the position of the thermometer can be shifted downward, or the upper or lower end of the cork can be cut away. The bulb should never be placed above the side-arm, since it is essential that it be covered completely by the vapor during the distillation. 18. Heating the Flask. - The best way of heating the distilling flask is determined by the boiling-point of the liquid to be distilled. If the liquid has a low boiling-point, up to about 80° or 90°, the flask should be placed in a water-bath in such a position that the level of the water is just below that of the liquid in the flask. Toward the end of the distillation the flask should be raised in order to prevent superheating the vapor of the liquid. With very volatile liquids great care is necessary to prevent this superheating. Another method which is often used is to place the flask on an asbestos board in which a hole is bored having a diameter about one-half that of the flask. The smallest flame which will furnish heat enough to boil the liquid is used. This method can be used for distilling in general, whatever the boiling-point of the liquid. If a flask of 250 cc. capacity or greater is used, it is advisable to support it on a wire gauze. This precaution is also advisable when the burner is put in place, and the distillation allowed to take place of itself. It is often better to hold the burner in the hand and keep the flame in motion during the distillation. In this way the process is more carefully watched and the rate of distilling can be controlled. The heating of the flask should be discontinued before all of the liquid has distilled; it is customary to leave a residue of 2 to 5 cc. in the flask. 19. Rate of Distillation. - The distilling flask should be heated in such a way that the distillate falls in drops from the end of the condenser at the rate of about one drop per second. Care should be taken to avoid the rapid distillation of very volatile, inflammable liquids, such as ether, alcohol, and carbon disulphide. If such liquids are distilled very rapidly, a part of the vapor is not condensed, and a fire may result when this vapor comes in contact with a near-by flame. In order to prevent accidents the method of collecting such liquids which is described in §34, page 23, should be used. 20. Distillation of High-boiling Liquids. - When a liquid boils above 150° an "air-condenser" should be used instead of the kind shown in Fig. 2, which is supplied with a water-jacket. If one of the latter type is used, the inner tube, cooled by running water, is apt to crack when the vapor of the high-boiling liquid comes in contact with it. The inner tube without a jacket is used as an air-condenser. When a substance which boils at a high temperature (above 300°) and solidifies readily is distilled, it is customary to use no condenser, but to collect the distillate directly at the end of the side-arm of the distilling flask. If, in this case, or when an air-condenser is used, the distillate solidifies before it reaches the receiver, the tube should be gently heated by passing the flame of a burner slowly along its length. It is necessary to prevent the filling of the side-arm of the flask with solid; if this occurs and boiling is continued, the vapor produced soon reaches a sufficient pressure to cause an explosion. When this method is unsatisfactory on account of the high melting-point of the substance, it is advisable to distil from a retort. On account of the large diameter of the neck of the retort, a considerable quantity of the solid can be collected in it. Before the solid fills the neck at any point, the distillation is stopped, the neck of the retort is heated, and the liquid collected in a beaker; the distillation is then continued. 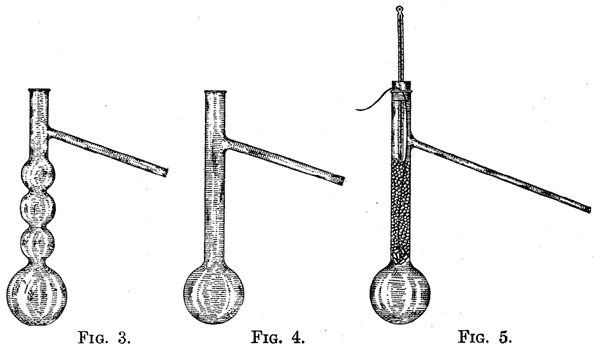 |
|||||||||||||||||||||||||||||||||||||||||||||
21. Fractional Distillation. - When it is necessary to separate two or more liquids by distillation, special forms of distilling flasks should be used. These are so constructed that they decrease materially the time required to effect a separation. This is accomplished by subjecting the vapor to gradual cooling before it is finally condensed. In this way the less volatile constituents of the vapor are condensed and returned to the flask, while the more volatile parts pass on through the condenser. The types of flasks used are illustrated by figures 3, 4, and 5. The arrangement represented in Fig. 5 is very efficient, especially when a small amount of a liquid is to be fractionated. After the liquid has been placed in the flask, a number of glass beads tied together with a cotton thread are supported by the thread, and the neck of the flask is filled to the place indicated in the diagram with glass beads. 22. The more complicated arrangements are supplied as tubes which are fitted by a cork to a round-bottomed flask. Figures 6, 7, 8, and 9 illustrate the forms commonly used. The most efficient form is that of Hempel, Fig. 9, which consists of a tube filled with glass beads. The least efficient form is that of Wurtz, Fig. 6. The efficiency of the Lebel-Henninger tube, Fig. 7, and of the Glinsky tube, Fig. 8, lies between the two extremes stated. 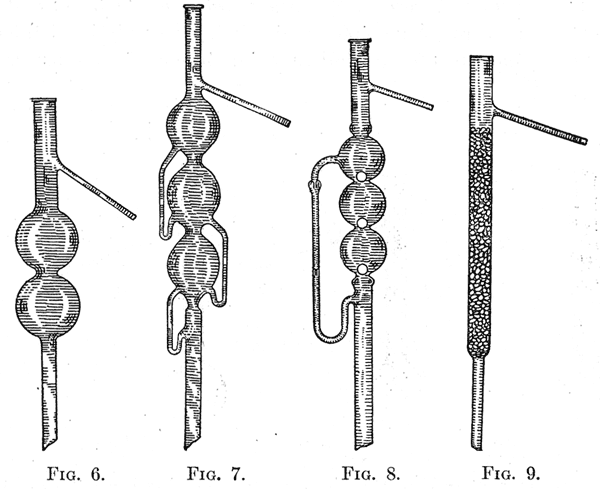 |
|||||||||||||||||||||||||||||||||||||||||||||
23. When a mixture of two liquids which boil at different temperatures is distilled, the temperature of the vapor during the distillation rises, in most cases, from the boiling-point of one of the liquids to that of the other. The distillate which is collected first contains a large proportion of the lower boiling liquid, while that collected toward the end of the operation is rich in the higher boiling liquid. In order to separate the two, the mixture is subjected to what is called fractional distillation. The process is carried out in the following way: The mixture is distilled slowly, and the receiver in which the distillate is collected is changed from time to time, as the boiling-point of the liquid rises. In this way the mixture is separated into what are called fractions. The number of fractions collected, and the limits of the boiling-point of the various fractions, are determined by the difficulty of separating the mixture and the purity of the products desired. The lowest boiling fraction is next placed in a clean flask and distilled. When the temperature reaches that of the upper limit of the fraction, the heating is stopped, and the second fraction added to the flask. Distillation is then continued until the upper limit of this fraction is reached, the distillate being collected in the appropriate receiver. The process is continued in this way until all the fractions have been distilled a second time. It will be found as a result of this fractionation that the distribution of the liquid in the several fractions is different from that obtained the first time. The fractions which boil at temperatures near those of the boiling-points of the constituents of the mixture increase in volume. By repeating the process a sufficient number of times, practically all of the liquid can be separated into its constituents. In the following table are given the results of the fractional distillation of a mixture of 50 cc. of methyl alcohol and 50 cc. of water. The volumes of the fractions obtained after each of six fractionations are recorded. |
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
When the liquids form a constant-boiling mixture, they can not be separated in pure condition by fractional distillation. The boiling-point of a mixture of ethyl alcohol and water, which contains 96 per cent by weight of the former, is lower than that of pure alcohol. As a consequence, when a mixture of the two substances is subjected to repeated fractional distillation, the constant-boiling mixture is obtained. In order to prepare pure alcohol it is necessary to remove the water from the mixture by chemical means. Very few cases of this kind are met with in the purification of organic compounds. Distillation Under Diminished Pressure 24. Many substances which decompose when distilled at atmospheric pressure, distil without decomposition when the pressure is reduced. This results from the fact that the temperature at which a substance boils is markedly affected by the pressure. For example, benzophenone boils at 306° at 760 mm. pressure, and at 170° at 15 mm. pressure. The effect of change in pressure on the boiling-point increases rapidly as the pressure decreases. Stearic acid, for example, boils at 291° at 100 mm., at 232° at 15 mm., and at 155° under the best vacuum obtainable with a mercury pump. A difference of 85 mm. in pressure from 100 mm. to 15 mm. causes a change in boiling-point of 59°, whereas a difference of 15 mm. from 15 mm. to O mm. lowers the boiling-point 77°. Many substances which distil with partial decomposition at atmospheric pressure can be distilled unchanged at the pressure which can be obtained with a good water-pump. A convenient arrangement of the apparatus required for distillation under diminished pressure is represented by Fig. 10. The flask to contain the substance to be distilled is fitted with a thermometer and a tube (a) which is drawn out to a fine opening at one end; to the other end of the tube is attached a piece of rubber tubing carrying a screw-clamp (b). This tube is provided to prevent violent bumping during the distillation. By regulating the screw-clamp after the apparatus has been attached to the vacuum-pump, a rapid stream of air-bubbles can be drawn through the liquid. As the latter is heated the vapor formed passes into the bubbles, and superheating and the consequent "bumping" are largely avoided. The position of the tube is so adjusted that the fine opening almost touches the bottom of the flask. 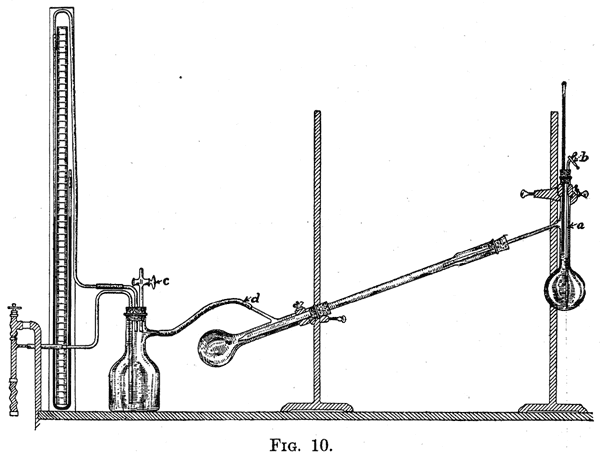 |
|||||||||||||||||||||||||||||||||||||||||||||
25. Other modifications of the form of the tube to admit air into the flask are often used. If the neck of the flask is small and it is impossible to insert into it both the thermometer and a glass tube of the ordinary diameter, the part of the tube which is to pass through the cork is drawn out to a capillary, and is inserted through a small hole made with a stout needle or the end of a file. One end of the tube is left with such a diameter that the rubber tubing and screw-clamp can be attached to it. 26. A second modification is often used on account of its convenience. It is illustrated in Fig. 11. A straight glass tube is selected of such a diameter that the thermometer passes into it easily. The tube is drawn out at one end to a small opening; it is then cut off at the other end at such a point that when the thermometer is placed in it and it is put into the flask, the bulb of the thermometer is in the correct position with regard to the side-arm of the flask. A piece of rubber tubing provided with a screw-clamp is attached to the upper end of the tube.  When liquids which boil at a very high temperature are distilled, it is customary not to use an air-condenser, but to connect the receiver directly with the side-arm of the flask which contains the liquid to be distilled.
When liquids which boil at a very high temperature are distilled, it is customary not to use an air-condenser, but to connect the receiver directly with the side-arm of the flask which contains the liquid to be distilled.In assembling the distilling apparatus, a rubber stopper may be used to attach the receiver to the condenser, provided care is taken to prevent the hot liquid from coming in contact with the rubber. Great care should be exercised in selecting the corks to be used; these should be as free as possible from holes. After the apparatus has been set up, small leaks can often be closed by painting the corks with collodion. A convenient arrangement of the manometer and the connections between the pump and the receiver are represented in Fig. 10. A number of forms of manometers are used to register the pressure inside the apparatus. A simple form which can be readily made from a supply of mercury, a meter-stick, and a piece of glass tubing is illustrated in the figure. In order to determine the pressure in the apparatus the readings on the scale opposite the levels of the mercury in the manometer are subtracted, and this difference is subtracted from the height of the barometer. It is necessary to insert an empty bottle between the pump and the receiver. When the apparatus has been evacuated, water may run back from the pump into the receiver as a result of a slight change in the pressure of he water caused by the opening of a cock in the neighborhood. The inserted bottle serves as a trap to catch this water. At c is a stopcock through which air can be let into the apparatus. This is of value at the end of a distillation, or in case the boiling liquid begins to froth or bump violently. In the latter case letting in a little air prevents the ejection of a part of the contents of the flask into the receiver. |
|||||||||||||||||||||||||||||||||||||||||||||
27. Method of Distillation. - Before introducing the liquid to be distilled, the whole apparatus should be tested. The screwclamp b should be closed, and the receiver connected by a heavy-walled tube to the pump. In no part of the apparatus should rubber tubing be used which collapses under diminished pressure. If no heavy tubing is available the connections can be made with glass tubing joined by ordinary rubber tubing, the ends of the glass tubes being brought together so that the connecting rubber tubes can not collapse. When the apparatus has been connected with the pump the pressure should be reduced to about 20 mm. If this can not be done, either the pump is a poor one, or the apparatus has not been well put together. The cause can be determined as follows: First, test the pump by connecting it directly with the manometer, making sure that there is not a leak in the connecting tubes. Second, disconnect the tube at d, and close it with a pinch-cock or glass rod. If the reduction in pressure is sufficient it will show that all the connections from the pump up to this point are tight. Next, disconnect the flask from the condenser and connect it by means of the side-arm to the pump and manometer. This will determine whether the cork provided with the thermometer and the tube to admit air is tight. It is probable that the leak will be found at this point. If everything is tight, connect the flask with the condenser, and the lower end of the latter with the pump. This will test the tightness of the joint between the flask and condenser. The apparatus is next completely adjusted, and tested again, when, if no leak has been discovered up to this point and the pressure can not be sufficiently reduced, it is evident that the connection between that condenser and receiver is at fault. When the apparatus has been found to be tight, the product to be distilled is introduced into the flask, which should be filled to not more than one-half its capacity. The suction is applied and the screw-clamp b opened very slowly so that a stream of air-bubbles passes through the liquid. The flask is heated by a bath containing oil or, preferably, a low-melting alloy such as Rose's or Wood's metal. It is often better to heat the flask by means of a free flame as we amount of heat applied can be quickly regulated. When a free flame is used it should be kept in constant motion, and the surface of the liquid where it comes in contact with the flask should be heated rather than the bottom of the flask. This can be done by moving the flame around the flask and letting it come in contact with the latter at the side and not the bottom. If frothing suddenly begins and there is a chance of the contents of the flask rushing over into the receiver, such a result can be prevented by opening the cock c which admits air to the apparatus. Special forms of apparatus have been devised to fractionate a liquid by distillation under diminished pressure. It is often simpler to use the apparatus described above, and change the receiver when the limits of the fractions have been reached. Distillation with Steam 28. Substances which are practically immiscible with water and have an appreciable vapor pressure at 100°, can be readily separated from those which have a very small vapor pressure at this temperature by passing steam through the mixture. The process which is of special value in separating organic compounds from tarry materials found in their preparation, is carried out in an apparatus arranged as represented in Fig. 12. The flask a is connected with a supply of steam; this can be conveniently generated in a kerosene can, which is supplied with a long glass tube reaching to the bottom of the can, to act as a safety-valve. Into the flask is put the substance to be distilled. The flask should be set up at an angle as indicated in the diagram. By placing the flask in this position any of the liquid which is violently thrown up against the flask as the result of the inrush of steam, will not be forced through the condenser into the receiver. The tube through which the steam is led should be so bent that its end almost touches the lowest point of the flask in its inclined position. By this means the steam is forced through the heavy liquid to be distilled, which is consequently kept in motion. If the liquid is not stirred up by the incoming steam distillation takes place very slowly. 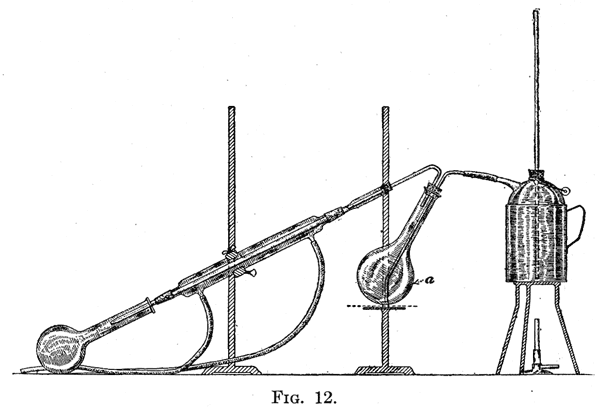 |
|||||||||||||||||||||||||||||||||||||||||||||
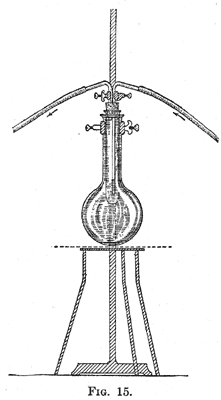
29. The vapor issuing from the flask consists of a mixture of steam and the volatile substance to be distilled. When this is condensed, two layers are formed. The theory of the process is briefly as follows: When a mixture of two immiscible liquids is heated, each substance vaporizes independently of the other. When the sum of the vapor pressures of the two liquids is equal to the pressure of the atmosphere, the mixture distils. The relation between the weight of the two substances obtained is determined by their molecular weights and their vapor pressures at the temperature of distillation. The case of nitrobenzene and water is an example. When steam is passed into nitrobenzene the mixture boils at 99°, when the atmospheric pressure is 760 mm. At this temperature the pressure of water vapor is 733 mm., and of nitrobenzene 27 mm.; consequently the relation between the weight of water and that of nitrobenzene is as 18 X 733 is to 123 X 27, or approximately 4 to 1. Although the vapor pressure of nitrobenzene is small at 99°, its large molecular weight compared with that of water leads to the result that about one-fifth of the product obtained by distillation with steam consists of nitrobenzene. When the vapor pressure of a compound is as low as 10 mm. at 100°, it can be advantageously distilled with steam. Orthonitrophenol can be conveniently separated from paranitrophenol on account of the fact that the former has an appreciable vapor pressure at 100°, and is consequently volatile with steam. 30. When the vapor pressure of a substance increases rapidly near 100°, the rate at which it distils can be markedly increased by adding to the mixture of it and water a substance soluble in water; the latter increases the boiling-point of the liquid. By saturating the water with calcium chloride a marked rise in the temperature at which distillation occurs can be effected, with the consequent increase in the vapor pressure of the organic compound undergoing distillation. When the vapor pressure of a substance is appreciable only at a temperature considerably above the boiling-point of water, it may often be separated from less volatile compounds by distillation with superheated steam. In this case the flask containing the substance is heated in an oil-bath, and steam which has been passed through a hot coil of copper is conducted through it. Extraction By extraction is meant the process of removing from a mixture, usually an aqueous solution, one or more substances by shaking with a liquid in which the substances to be removed are soluble. Aniline, for example, is somewhat soluble in water; when the solution is shaken with ether a large part of the aniline is removed from the water and passes into solution in the ether. As aniline can be recovered much more readily from an ethereal solution than from an aqueous solution, extraction of such solutions is made use of in the preparation of the compound. The liquid used for extracting must be immiscible with the solution to be extracted. 31. Method of Extraction. - In extracting a solution it is shaken in a separatory funnel with a liquid in which the substance to be extracted is readily soluble. The substances commonly used for this purpose are ether, chloroform, benzene, petroleum ether or ligroin, and carbon disulphide. Ether is generally used as it is an excellent solvent for many organic compounds, and, on account of its low boiling-point, it can be readily removed. The disadvantages connected with the use of ether are its great inflammability and the fact that it is somewhat soluble in water and dissolves appreciable quantities of water. Water dissolves approximately 10 per cent of its volume of ether. When large volumes of aqueous solutions are extracted there is a loss of ether, which is an expensive substance. This loss is decreased by saturating the solution to be extracted with sodium chloride. On account of the fact that ether dissolves about 2 per cent of its volume of water, ethereal extracts have to be dried, in most cases, before the ether is removed by evaporation. 32. The relation between the volume of an aqueous solution to be extracted and the volume, of the solvent used for extraction, is determined by the relative solubilities of the substances to be extracted in water and the solvent used. If an aqueous solution of a substance is extracted with ether, the amounts of the substances found in the two liquids will be proportional to the solubilities in the two solvents and to the amounts of the latter. If the substance is equally soluble in water and in ether, and the volumes of the two liquids are the same, after extraction one-half of the substance will be found in the ether. If the substance is twice as soluble in ether as in water, the relation of the amount present in the ether will be to that present in the water as two is to one, that is, two-thirds will be present in the ether and only one-third in the water. By shaking the aqueous solution with a second portion of ether, two-thirds of the substance present, that is two-thirds of one-third, or two-ninths, of the original amount will be removed and one-ninth will remain dissolved in the water. After three extractions but one twenty-seventh of the substance will remain dissolved in the water. In the above example a certain volume of a solution was extracted three times, using each time a volume of ether equal to that of the aqueous solution. The result would have been different if the solution. had been extracted with the three volumes of ether in one operation. In this case the substances would have been divided between the ether and the water in the ratio of 3 X 2:1; that is, one-seventh would have remained dissolved in the water. As the result of extracting the solution with the same volume of ether in three operations using one-third of the solution each time, but one twenty-seventh remains dissolved in the water. It is evident, therefore, that the most efficient way to extract a substance is to shake the solution a number of times with small amounts of the extracting agent. 33. The relation between the volumes of the extracting liquid and of the solution, and the number of times the solution should be extracted, vary widely with the relative solubility of the substance to be extracted. In general, in the case of a substance which is much more soluble in ether than in water, three extractions will be sufficient if a volume of ether equal to about one-fourth of that of the aqueous solution is used. In order to determine whether the extraction has been carried far enough, a sample of the last ethereal extract should be evaporated on a watch-glass on a steam-bath. The amount of the residue will determine whether a fourth extraction is desirable. If a large volume of liquid is to be extracted, and a separatory funnel of appropriate size is not available, the liquid can be placed in a flask and shaken with ether; the major part of the latter can be decanted off, and the rest separated in a small separatory funnel. 34. Separation of the Extracted Substance. - If the substance is a solid, and a small amount of the extracting liquid has been used, the solution can be evaporated to dryness and the residue crystallized. If it is desired to recover the ether or other solvent used in the extraction, the solution should be placed in a flask and the solvent distilled off on a water-bath as described below. When the substance to be obtained from the solution is a liquid which is to be finally distilled, it is necessary to dry the extract before the removal of the solvent. The drying agent must be selected according to the principles stated in §36, page 24. If ether has been used, the solution should not be set aside to dry in a thin-walled flask which has been stoppered; sufficient heat is at times generated as the result of the union of the water and the drying agent to break the flask. A bottle or distilling flask should be used. It is not advisable to place the extract in a beaker or other open vessel, as the solvent will be lost if the solution stands for some time. When the solution is dry, the solvent can be removed by distillation. If ether or any other very volatile and inflammable liquid is used, the flask containing the solvent should be heated on a water-bath, and should be provided with a long water-jacketed condenser and a special form of receiver. This is made by attaching to the condenser, by means of a tightly fitting stopper, an adapter, which, in turn, is attached in the same way to a filter-bottle. The side-arm of the latter is provided with a rubber tube of such length that it reaches nearly to the floor. By taking these precautions accidents caused by fires are prevented, as the only way in which ether vapor can escape from the apparatus is through the rubber tube; as ether vapor is very heavy and as any which escapes is delivered at the level of the floor, there is little chance of its being ignited by any flames on the laboratory desks. The receiver should be dry in order that the ether which distils over may be used in transferring the residue after the distillation to a flask of appropriate size for the final distillation. The dry ethereal extract is decanted, or, better, filtered from the drying agent into the distilling flask, great care being taken to prevent any heavy aqueous layer from getting into the flask. Such a layer is frequently formed when potassium hydroxide is used as a drying agent; the compound extracts water from the ether and forms a saturated aqueous solution. The flask is next tightly corked, attached to the condenser and receiver, and the ether distilled off on a waterbath. When all the ether has evaporated the residue is poured into a flask of appropriate size for distillation. As an appreciable amount of the substance adheres to the larger flask from which the etheral extract was distilled, it should be washed out twice with a few cubic centimeters of the ether which have been distilled off, and these washings added to the flask from which the final distillation is to be made. In this distillation the flask should be heated slowly at first until the small amount of ether has been driven out. The final distillation should be made as described in §15. 35. Sublimation. - This process is of special value when it is desired to separate a solid which is volatile from substances which do not vaporize readily. It generally yields a very pure substance, but it often leads to loss. The process is most easily carried out between two watch-glasses which fit closely. The substance which has been carefully dried is placed in one of the glasses. This is covered with a piece of filter-paper, in which a few small holes have been cut to allow the passage of the vapor. The second glass, placed in an inverted position, is fastened to the first by means of a specially constructed clamp. The apparatus is heated slowly on a sand-bath and the upper watch-glass is cooled by putting on it pieces of filter-paper which are kept moist with cold water. It is necessary to keep the upper watch-glass at a temperature lower than the melting-point of the substance to be sublimed. 36. Drying Agents. - Many organic substances are prepared in water solution or in their preparation are washed with water to remove soluble impurities. In this case it is necessary to dry them, if they are liquids, before they are distilled. It is necessary to select as a drying agent a substance which does not react with the compound to be freed from water. The substance generally employed is anhydrous calcium chloride, which is commonly used in a granular form; for many purposes the chloride which has been fused in the form of sticks is preferable. Calcium chloride forms addition-products with hydroxyl compounds and should not be used as a drying agent for alcohols, phenols, etc. Alcohols are commonly dried with quicklime. Water is usually removed from basic substances by treating them with solid potassium hydroxide. Anhydrous copper sulphate is an excellent drying agent for most substances; another salt, anhydrous sodium sulphate, is frequently used. It must be remembered in the latter case that hydrated sodium sulphate loses its water of crystallization at 33°; it is evident that the salt acts as a drying agent only below this temperature. The most powerful drying agents are sodium and phosphorus pentoxide. The use of the former is evidently limited to those substances which do not react with the metal. Sodium is used to remove the last traces of water only, the substances being previously dried with calcium chloride which removes most of the water. 37. The drying agent should remain in contact with the substances to be dried for from 2 to 3 hours, if the two are left in contact at room temperature. If convenient, the mixture should be set aside over night. Whether the drying agent should be removed from the substance before it is distilled, is determined by the boiling-point of the substance and the stability of the compound formed between the water and drying agent. It is advisable, however, except in the case of substances which boil below 80°, to remove the drying agent before distillation. The last traces of water may be removed from liquids which boil above 200° by drawing a current of air through them while they are gently heated. 38. Use of the Reflux Condenser. - It is often necessary to heat together two or more substances for a number of hours. If all the substances boil above the room temperature an open vessel provided with a reflux condenser is used. The arrangement of the apparatus is shown in Figs. 13 and 14. If it is desired to distil the product formed directly from the flask, the arrangement represented in Fig. 14 may be used. In this case the side-arm of the distilling flask is covered with a cork into which a hole has been bored half through its length. The side-arm is so placed that the liquid which condenses in it returns to the flask. Heating with a reflux condenser is usually carried out in a round-bottomed flask as shown in Fig. 13. If the contents are apt to boil with bumping the flask is usually heated on a sandbath; otherwise a free flame and a wire gauze are used. If the liquid boils above 150° an air-condenser is used instead of a condenser provided with a water-jacket. When it is necessary to heat with a reflux condenser substances which destroy cork and rubber, a simple device can be used which is represented in Fig. 15. A test-tube is selected which fits loosely into a long-necked, round-bottomed flask. The tube is supplied with a rubber stopper and tubes as shown in the drawing. Water is passed through the tube, which is supported in the neck of the flask by means of a clamp. In many syntheses hydrogen chloride or bromide is given off. It is inadvisable to let these gases get into the room even when the apparatus is placed under a hood. The top of the condenser should be provided with a tube bent at two right angles. This tube should reach to about 1 inch of the surface of water contained in a flask supported by a ring-clamp to the stand to which the condenser is attached. If it is necessary to keep the contents of the flask dry during the reaction, a straight drying tube containing calcium chloride should be inserted between the condenser and the flask containing the water. 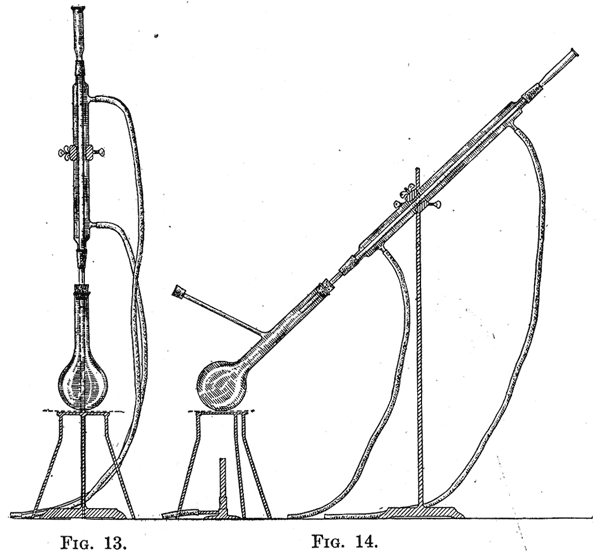 |
|||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||
39. Prevention of Bumping. - When a substance boils irregularly and "bumps," even boiling can often be obtained by placing in the vessel a few pieces broken from a porous plate. The so-called capillary boiling tubes are also valuable. They can be made by drawing out pieces of glass tubing to stout capillary tubes; these are cut off at such a length that they will reach from the bottom of the flask to well into the neck. They are next put into the flame and melted at such a point that the tube tuses together at about 0.5 cm. from one end. When this end is placed under a liquid the small cavity is filled with air, and as the liquid boils bubbles of vapor are formed at the end of the tube and even boiling results. If the liquid cools below its boiling-point after it has been heated some time, the cavity at the end of the boiling tube becomes filled with liquid as the result of the condensation of the vapor. In this case the tube must be withdrawn and the drop of liquid shaken from it, or a new tube must be inserted.  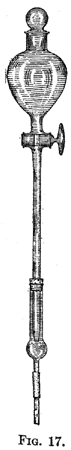 40. Dropping Funnels. - A dropping funnel like that shown in Fig. 16 is very useful. With a funnel of this type it is possible to observe at the point marked a the number of drops which pass through the funnel. If such a funnel is not available one can be made from an ordinary separatory funnel by attaching to its end a short calcium chloride drying tube as shown in Fig. 17, and connecting the latter by means of a glass tube to the apparatus to be used. 40. Dropping Funnels. - A dropping funnel like that shown in Fig. 16 is very useful. With a funnel of this type it is possible to observe at the point marked a the number of drops which pass through the funnel. If such a funnel is not available one can be made from an ordinary separatory funnel by attaching to its end a short calcium chloride drying tube as shown in Fig. 17, and connecting the latter by means of a glass tube to the apparatus to be used. |
|||||||||||||||||||||||||||||||||||||||||||||
41. Manipulation of Sodium. - Sodium is used in the preparation of a large number of organic compounds. As the metal reacts rapidly with oxygen and with water-vapor, it should not be allowed to stay in contact with the air any longer than is necessary. When sodium is to be cut with a knife or pressed into a wire, the coating which covers the metal should be first carefully removed and rejected; and the sodium should be placed immediately under dry ether, if it is not to be used at once. In many preparations in which sodium is used a part of the metal is left unchanged at the end of the experiment. Great care must be exercised in getting rid of the residue. It should not be left in an unlabeled flask or bottle. Under no circumstances should the product be allowed to come into contact with water. Small quantities of alcohol should be added from time to time to the sodium, until enough of the liquid is present to dissolve the residue, or to make with it a thin paste. This mixture should then be poured slowly into water, in order to prevent an accident in case any sodium is enclosed within a mass of inert solid. When sodium is used in a preparation, great care should be taken to prevent water entering the vessel containing the metal, either as the result of using a poorly fitting cork or a defective condenser. The Manipulation of Small Quantities of Substances In the identification of organic compounds it is generally necessary to transform them into other compounds, the properties of which are determined as an aid in the identification. It often happens that but a few grams of a substance are available for the purpose. The successful handling of such small quantities requires careful work, and the student should have opportunity to learn the special technique required. 42. Crystallization. - In the purification of small quantities of substances by crystallization a solvent should be selected in which the substance is more or less difficultly soluble, and care should be taken to avoid an excess of the solvent. It is best not to add enough of the solvent to completely dissolve the substance, even at the boiling temperature. The hot solution should be rapidly filtered. This can be done best under diminished pressure. A filter-bottle is selected of such a size that it will hold a 6-inch test-tube and permit a funnel being placed in the neck of the bottle in the usual way. The funnel is fitted with a perforated plate and a circular piece of filter-paper which is cut with a diameter about 8 mm. greater than the plate. The paper is put in place and pressed down so that it covers the joint between the plate and the funnel. A little of the liquid is poured through the funnel and the suction applied. This serves to set the paper firmly in place. The liquid is poured out of the test-tube, which is then replaced in the bottle. The funnel is put in place and the solution to be filtered is poured into it. Under the diminished pressure, the solution filters rapidly before the compound in solution can crystallize out. The method of filtration described in §10, page 6, is also to be recommended. 43. A filter-bottle provided with a perforated plate and test-tube is used to separate crystals from the mother-liquor containing them. While connected with the suction-pump the mixture of crystals and mother-liquor is poured slowly down a glass rod onto the filter-paper. If a very small amount of crystals is to be separated, care should be taken to collect them in a single spot and not to spread them out over the entire plate. In this way the crystals can be collected in a small mound, a few millimeters in diameter, which can be readily removed from the paper when dry. The crystals should be washed as directed in §12, page 7. 44. Distillation of Small Amounts of Liquids. - The liquid should be distilled from a 5 cc. distilling flask. If this is not available the neck of a broken distilling flask can be converted into a serviceable piece of apparatus for this purpose by sealing it at a point about 5 cm. from the side-tube. In distilling with small flasks an asbestos shield as described in §18, page 10, and a very small flame should always be used; a short thermometer reduces the error arising from stem-exposure. The boiling-point of 1 cc. of liquid can be determined in this way. In heating the substance, the flame should be applied, at first, in such a way that the vapor condenses in the flask just before it reaches the side-tube. In this way the thermometer is heated up to the temperature of the vapor before the latter is driven over. If the distillation is carried on rapidly the small amount of liquid will have distilled over before the thermometer has been heated up to the temperature of the vapor. For a method of determining the boiling-point of very small quantities of a liquid see Smith and Menzies, J. Am. Chem. Soc., 33, 897. 45. If it is necessary to distil fractionally a small amount of liquid, a flask should be selected the side-arm of which is as far away from the bulb as possible (see Fig. 4, page 11). Such a flask can be furnished with an efficient fractionating column as follows: Glass beads which pass snugly into the neck of the flask are tied to the end of a cotton thread, or if necessary a fine platinum wire. After the liquid has been introduced, the beads are hung at the bottom of the neck of the flask, which is then filled with more beads, just enough room being left for the thermometer. A column of beads only a few centimeters high is remarkably efficient in bringing about the separation of liquids of different boiling-points through distillation (see Fig. 5, page 11). 46. Extraction of Small Amounts of Substances. - When a small amount of substance is to be extracted the solution is shaken with about an equal volume of ether or other solvent in a test-tube. To separate the two liquids the upper layer is drawn into a pipette. In order to be able to see clearly the position of the end of the pipette, a long rubber tube is connected with it so that the test-tube may be held at the level of the eye while the liquid is being drawn up. If a number of extractions are to be made a simple apparatus which can be made readily is of value. This consists of a test-tube with a side-arm placed at a point slightly above the middle of the tube. The tube is filled with the liquid to be extracted up to the side-arm, which is closed by the end of the forefinger. Ether is added, the tube closed with the thumb, and the mixture shaken. A vessel to hold the ether is placed under the side-arm; when the finger and thumb are removed the ether runs out of the tube. 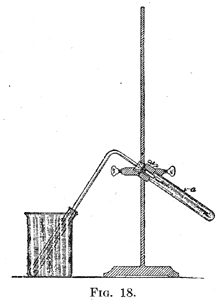 47. Preparation of Liquids on the Small Scale. - It is often necessary to prepare small amounts of liquids, which are obtained as the result of heating together two or more substances, and subsequent distillation. An example is the preparation of ethyl iodide by heating alcohol and hydriodic acid. A simplified form of apparatus for such work is shown in Fig. 18. 47. Preparation of Liquids on the Small Scale. - It is often necessary to prepare small amounts of liquids, which are obtained as the result of heating together two or more substances, and subsequent distillation. An example is the preparation of ethyl iodide by heating alcohol and hydriodic acid. A simplified form of apparatus for such work is shown in Fig. 18.The test-tube a is supported by a clamp; the test-tube which serves as a condenser is surrounded by cold water contained in a beaker. It is important that the tube from a should extend nearly to the bottom of the condenser, in order that any vapor which passes over may come in contact with the walls of the tube, which are cooled by the water in the beaker. In using an apparatus like this, the distillation should be made very slowly. |
|||||||||||||||||||||||||||||||||||||||||||||
Determination of Physical Properties 48. Calibration of Thermometers. - The thermometers provided for use in determining the physical properties of organic compounds are usually far from accurate. If very exact measurements are to be made, a high-grade thermometer should be calibrated according to the methods described in books dealing with physical measurements. For students' work in organic chemistry, a cheap thermometer may be used provided it is calibrated with reasonable care. A thermometer which is accurate to within 0.5° is satisfactory. Although most thermometers register correctly at 0° and 100°, it is well to make a calibration at these points. To determine the reading at 0, a beaker of 100 cc. capacity is filled with finely chopped ice, and water is added until it reaches to within about 2 cm. of the surface of the ice. The thermometer is inserted into the ice and water until the zero point is just above the surface, so it can be seen. The reading on the thermometer is noted when the mercury no longer falls. Care should be taken to have the eye at the height of the top of the mercury column to avoid parallax. To calibrate the thermometer at higher temperatures it must be heated in such a way that the entire mercury thread is exposed to the vapor of a boiling liquid. This can be conveniently done as follows: Attach to an 8-inch test-tube by means of a closely fitting cork (not a rubber stopper) a short air-condenser. Place in the tube about 10 cc. of water, and support the tube and condenser by means of a clamp over a wire gauze. Attach a cotton thread to the thermometer, and lower it through the condenser until the bulb is about 4 inches above the liquid. The thermometer is held in place by tying the upper end of the thread around a small piece of glass rod, which is then placed across the end of the condenser. The water is boiled, and when the entire thermometer is in the vapor, the position of the mercury is noted. The thermometer should be calibrated at higher temperatures by determining the boiling-points of other liquids in the apparatus described above. The latter should be thoroughly dried and a fresh cork used to avoid the presence of water. The substances which can be used conveniently are aniline, naphthalene, and benzophenone. The boiling-points of these substances at 760 mm. pressure are, respectively, 183.7°, 218.1° and 306.1°. These are the temperatures recorded when the entire thread of mercury in the thermometer is exposed to the vapor of the boiling liquid. The barometric pressure should be noted and a correction made of 0.10 for each 2.7 mm. of pressure; if the observed pressure is less than 760 mm. the correction should be added, and if greater it should be subtracted. The substances used should be pure; the aniline must be redistilled, and the first part of the distillate rejected, in order to free it from the small amount of water that it usually contains. The thermometer should be marked with a number, and the corrections to be applied recorded, in the notebook. 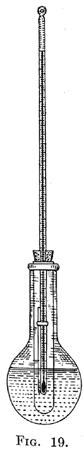 49. Determination of Melting-points. - A form of apparatus which can be conveniently used to determine melting-points is represented by Fig. 19. It consists of a 100 cc. round-bottomed flask and a test-tube which fits loosely into the neck of the flask. A thermometer is supported in the test-tube by a cork in which a slit has been cut so that the graduations of the former are visible. The flask and test-tube are filled with pure concentrated sulphuric acid to the heights indicated in the drawing. If the sulphuric acid becomes brown as the result of the introduction of traces of organic matter, a tiny piece of a crystal of potassium nitrate will destroy the color. 49. Determination of Melting-points. - A form of apparatus which can be conveniently used to determine melting-points is represented by Fig. 19. It consists of a 100 cc. round-bottomed flask and a test-tube which fits loosely into the neck of the flask. A thermometer is supported in the test-tube by a cork in which a slit has been cut so that the graduations of the former are visible. The flask and test-tube are filled with pure concentrated sulphuric acid to the heights indicated in the drawing. If the sulphuric acid becomes brown as the result of the introduction of traces of organic matter, a tiny piece of a crystal of potassium nitrate will destroy the color.The substance, the melting-point of which is to be determined, is dried, usually in the air, and is powdered by rubbing it with a spatula on a clean porous tile. It is then placed in a small capillary tube which has an internal diameter of about 1 mm. |
|||||||||||||||||||||||||||||||||||||||||||||
50. Tubes for the determination of melting-points are conveniently made as follows: A piece of glass tubing of 5 mm. internal diameter is softened by rotating it in a flame; it is removed from the flame and after 2 seconds is slowly drawn out so that the tube is elongated about 10 cm., and the tube formed has at its narrowest point an external diameter of about 1.5 mm. The tube is again heated and drawn out. The process is repeated until the tube is of such a length it can not be conveniently handled. The appearance of the tube so prepared is represented by Fig. 20. The distance from a to c should be about 15 cm. The tube is cut at a, c, e, etc. These pieces are then cut in the middle at b, d, etc., and each piece sealed at these points by holding in a flame. A number of tubes should be prepared at one time. In order to avoid getting dust into the tubes, they should be kept in a test-tube provided with a cork, or should be placed in a small beaker with the open ends downward. The substance to be melted is placed in a tube. This can be done readily by pressing a little of the substance into the open end of the tube with the aid of a spatula, grasping the tube by the closed end, and drawing a file lightly, over it. The vibrations produced in this way cause the powder to fall in the tube. The process is repeated until a layer of the substance from 0.5 to 1 cm. thick has been formed. The tube is attached to the thermometer by means of a rubber band which is made by cutting of a piece of tubing about 2 mm. in length from a rubber tube of such a diameter that it fits the thermometer snugly when drawn over it. This band should be placed at least 2 cm. above the surface of the acid.  |
|||||||||||||||||||||||||||||||||||||||||||||
51. In making a preliminary determination of an unknown melting-point, the flask is heated cautiously with a free flame in such a way that the thermometer rises at the rate of about ten degrees per minute. The temperature should be noted at which the substance first shows signs of melting and when it has completely liquefied. As the thermometer is rapidly rising, this result serves only as a guide for the determination of the melting-point. When the apparatus has cooled to a number of degrees below the temperature at which signs of melting were evident, a new tube containing another sample of the substance is introduced into the apparatus and the melting-point redetermined. All melting-point determinations should be made with samples which have not been melted. If the thermometer is at a high temperature when it is removed from the bath, it should not be cooled by placing it in cold water; if this is done the glass is apt to crack. The tube is heated rapidly until the temperature is at least ten degrees below that at which the substance melts. The flame should be removed and the temperature allowed to rise slowly. When the thermometer ceases to rise the flask should be heated cautiously so that when the flame is removed the temperature does not rise more than one degree. In the case of a substance which melts without decomposition, the temperature should be allowed to rise at the rate of about two degrees per minute for the last five degrees. An endeavor should be made to have such control of the bath that the thermometer can be forced up a degree at a time. The heating is continued until the substance completely runs down the tube as a liquid. The point at which this occurs and that at which the substance begins to form in droplets should be recorded as the melting-point of the sample studied. If these are separated by more than one degree, the substance is more or less impure. If the substance shows signs of contracting or softening before it begins to melt, the fact should be noted as it is an indication that it is impure. The determination of the melting-point should be repeated, and great care taken to allow the temperature to rise very slowly when the substance begins to melt. It may happen that the substance melts within one degree, but that it appears to melt over a considerable range of temperature on account of the fact that the heat has been applied so rapidly that the thermometer rises a number of degrees during the time required to melt the substance. When a substance appears to be pure and gives a sharp melting-point, a number of determinations should be made. If these give the same result, the melting-point as determined may be considered to be the correct one. If a substance does not melt sharply it should be recrystallized, and a second determination made. As a result of the purification the melting-point obtained will probably be higher than before. The process is repeated as long as the melting-point rises. 52. The melting-point determined as indicated is below the true melting-point of the substance on account of the fact that only part of the mercury is heated to the temperature of the melting substance. A close approximation of the true melting-point can be found by applying the correction which is found by substituting in the formula N(t - t') 0.000154, in which N is the number of degrees of mercury not heated directly by the acid in the bath, t the observed temperature and t' the average temperature of the stem outside the bath. The correction, which is added to the observed temperature, is, in the case of a bath like that described, and when the usual form of thermometer which registers up to 360° is used, about 1° at 100°, about 2° at 140°, 3° at 170°, and 6° at 220°. 53. Determination of Boiling-points. - The determination of the boiling-point of a substance is made during the final distillation in its purification. If 20 or more grams are distilled, the boiling-point as determined in the apparatus described above under distillation, may be considered to be correct. Care must be taken, however, not to distil all of the liquid, as when this is done the vapor toward the end of the distillation becomes superheated, and the thermometer does not register the true boiling-point of the liquid. 54. If but a small amount of the substance has been prepared, or the boiling-point of a small sample of an organic compound is desired, the determination is best made in the manner described below, which largely prevents superheating. About 10. cc. of the liquid is placed in a 15 cc. flask provided with a thermometer the bulb of which is placed just below the side-tube. The flask is supported on an asbestos board in which has been carefully cut by means of a sharp cork-borer of brass, a hole about 2 cm. in diameter. The bottom of the flask should fit the hole tightly so that the hot gases from the flame can not come in contact with the upper part of the flask. On account of the smallness of the flask, a large part of the thermometer is not heated to the temperature of the vapor and a correction should be made for stem exposure as described above under the determination of melting-points. In order to determine the mean temperature of the stem, a second thermometer should be attached by means of rubber bands to the one used to indicate the temperature of the vapors in the flask. The bulb of the second thermometer should be placed at a point half way below the upper end of the first thermometer and the cork. The condenser to be used is determined by the boiling-point of the liquid (see §20, page 11). The flask should be heated by a free flame of such a size that when it comes in contact with the lower part of the flask it furnishes sufficient heat to keep the liquid boiling at such a rate that about 0.5 cc. distils per minute. The distillation should be stopped when the level of the liquid is just below that of the asbestos-board that supports the flask. If a very accurate determination of a boiling-point is to be made, the substance should be distilled from a tube of such a length that the thermometer can be heated by the vapor up to the point where the top of the mercury column stands when the substance is boiling. When determined in this way no correction for stem exposure is necessary. 55. As the boiling-point varies with the pressure, in recording accurately determined boiling-points, both the pressure and the temperature are stated. When the determination is made at a pressure within 40 mm. of the normal pressure, 760 mm., a correction can be applied to the observed reading to reduce the boiling-point to that of the substance at the normal pressure. This correction is 0.1° for each 2.7 mm. pressure. If the observed pressure is less than 760 mm., the correction should be added; if greater, it should be subtracted. It is evident that this correction should not be applied if the substance does not boil sharply and if every precaution has not been taken to have a pure substance and to eliminate the errors which commonly are present in a boiling-point determination made in the ordinary way.  56. Determination of Specific Gravity. - The determination of specific gravity is often made in the identification of organic compounds. For this purpose results accurate to two units in the third decimal place are sufficient; these may be obtained by using as little as 1 cc. of a liquid. A convenient form of apparatus for the determination is described by Mulliken[1]. It consists of a 1 cc. pipette and a glass tube, closed at one end, into which the former passes freely (see Fig. 21). 56. Determination of Specific Gravity. - The determination of specific gravity is often made in the identification of organic compounds. For this purpose results accurate to two units in the third decimal place are sufficient; these may be obtained by using as little as 1 cc. of a liquid. A convenient form of apparatus for the determination is described by Mulliken[1]. It consists of a 1 cc. pipette and a glass tube, closed at one end, into which the former passes freely (see Fig. 21).When the apparatus is weighed, it is stood on the pan of an analytical balance and the loop of wire is placed over the hook which supports the scalepan. The apparatus is calibrated as follows: The pipette and tube are cleaned, dried, and weighed. The pipette is next filled to the mark with distilled water, the temperature of which is noted; any liquid adhering to the outside of the pipette is removed. The pipette is put into the tube and the water allowed to flow out into the latter. The apparatus is weighed again. The increase in weight is the weight of water, at the observed temperature, which fills the pipette. In determining the specific gravity of a substance the pipette is filled with it as before and weighed, and the temperature of the liquid is noted. The specific gravity is obtained by dividing the weight of the substance by the weight of the water. The result obtained is the specific gravity at the temperature at which the substance was weighed, compared with water at the temperature at which it was weighed. As it is customary to refer specific gravities to water at 4°, the volume of the pipette should be calculated for the weight of water which it contains. In the following table are given the weights of 1 cc. of water at temperatures from 14° to 30°. |
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
The volume of the pipette may be determined by dividing the weight of the water by the weight of 1 cc. of water at the temperature at which the water was weighed. In calculating the specific gravity of a substance the weight observed is divided by the volume of the pipette; the result is the specific gravity of the substance at the observed temperature compared with water at 4°. If the number determined in this way for a substance weighed at 18° was found to be 1.365, the result would be expressed as 1.365 18°/4°. If a number of determinations of specific gravity are to be made, it is advisable to calibrate the pipette to hold 1 cc. If this is done the observed weight of the substance which fills the pipette is its specific gravity referred to water at 4°. The Qualitative Analysis of Organic Compounds 57. Test for Metallic Elements. - About 0.1 gram of the substance is heated in a clean dry porcelain crucible. If it burns with a flame or leaves a residue of carbon it is probably organic. If the residue is black it should be heated until all the carbon has burned off. If the original substance contained a metal there will be left in the crucible after ignition either a metal, an oxide, or a carbonate. This residue should be identified by the methods of inorganic qualitative analysis. Organic compounds which leave residues on ignition are usually metallic salts of acids. If it is reasonably certain that the substance does not contain a metal which is readily reduced by carbon, the ignition can be made on a platinum foil. If there is doubt as to whether the compound is organic, a sample of it can be heated in a hard glass tube with powdered copper oxide, and the evolved gas tested for carbon dioxide. If the substance is volatile it is necessary to place a mixture of it and copper oxide in the bottom of the tube and then add enough of the oxide to make a layer at least 5 cm. thick. The tube is carefully heated, beginning at the point farthest from the bottom of the tube. 58. Test for Non-metallic Elements. - The substance to be analyzed is first decomposed by heating it with metallic sodium, and the resulting mixture, which may contain the following compounds of sodium, is analyzed: chloride, bromide, iodide, phosphide, sulphide, cyanide, and sulphocyanide. The decomposition is accomplished as follows: A clean, dry 6-inch test-tube is supported near the open end in a vertical position by means of a clamp and iron stand. A piece of sodium equal in size to a cube 3 mm. on each edge is cut and wiped free from oil by means of a filter paper. Any deposit on the sodium should be rejected, and the piece selected should have clean freshly cut surfaces on all sides. The test-tube is warmed, and the sodium dropped in. The burner is placed directly under the tube which is heated until a layer of the sodium vapor about 1 cm. thick is formed. The substance to be analyzed is now dropped into the tube, care being taken to have it fall directly onto the hot sodium without coming into contact with the tube. If the substance is a liquid, about 3 drops should be used. If it is a solid, a little should be taken up on the end of a pen-knife or spatula and dropped into the tube; second portion should then be added. The flame is removed immediately, and when the tube is cold, the lower end of it should be broken off by tapping the tube with a pestle in a clean, dry mortar. If, during the heating, some of the undecomposed substance has settled on the upper part of the tube, this part should be rejected; only the very end which contained the sodium vapor should be used. About 2 cc. of alcohol is then added to react with any excess of sodium present. When the metal has ceased acting, the contents of the mortar are ground with 20 cc. of distilled water, transferred to a small beaker, heated to boiling, and filtered. If the decomposition has been satisfactorily accomplished a colorless solution is obtained. If this is not the case, a second fusion with sodium should be made, and great care taken to introduce the substance directly into the sodium vapor. Separate portions of the filtrate are tested for non-metallic elements as follows: Test for Sulphur. - Add to about 2 cc. of a dilute solution of sodium hydroxide 2 or 3 drops of a solution of lead acetate. To the resulting mixture add about 5 cc. of the filtrate obtained as described above. If sulphur is present, black lead sulphide will be formed. If but a small amount of sulphur is present, a yellow color may be produced without the formation of a precipitate. If this is the case, filter the solution after a few minutes, and examine the paper for a precipitate of lead sulphide. 59. Test for Nitrogen. - Boil for a minute about 2 cc. of the solution from the fusion with 5 drops of a 10 per cent solution of sodium hydroxide and 5 drops of a solution of ferrous sulphate. Cool, and add dilute hydrochloric acid drop by drop, until the solution becomes acid and the precipitate of ferrous hydroxide has dissolved. An excess should be avoided as the reaction which takes place if nitrogen is present-the formation of ferric ferrocyanide - is more delicate in the absence of an excess of acid. If a large amount of acid appears to be necessary, it is evident that the original fusion did not completely destroy the organic compound. If a blue or green color does not develop on adding the acid, a drop of ferric chloride should be added to the tube. It often happens that enough ferric ferrocyanide is not formed to produce the characteristic blue precipitate; the formation of a clear green solution is ample proof of the presence of nitrogen. Such a solution usually deposits a blue precipitate on standing. 60. Test for Halogens. - A preliminary test for halogen should be made with some of the original substance. The so-called Beilstein test is made as follows A piece of stout copper wire is bent around the end of a lead pencil to make a small loop at one end. This end is then heated in a Bunsen flame until it no longer imparts a color to the flame. The wire is allowed to cool, and the clean end is inserted into the substance to be tested if it is a liquid, or a bit of it, if solid, is supported in the loop on the wire. It is then put into the flame. If a halogen is present a green color is imparted to the flame. The test should be repeated a number of times in order to have the conclusion definite. If a halogen is found to be present the solution obtained from the decomposition with sodium should be tested. If sulphur and nitrogen have been found to be absent, about 1 cc. of the solution is acidified with nitric acid, and silver nitrate is added. The color of the precipitate formed is an indication of the halogen present, provided but one is present. If the presence of bromine or iodine is suspected, a second portion of the original solution is acidified, and about 2 cc. of carbon disulphide is added; chlorine water or a solution of sodium hypochlorite is then added, drop by drop. If either sulphur or nitrogen is present, it is necessary to boil the original solution for several minutes with a few drops of dilute sulphuric acid to remove hydrogen sulphide and hydrocyanic acid before the tests for the halogens are applied. 61. Test for Phosphorus. - One cubic centimeter of the original solution from the fusion is boiled for 1 minute with 3 cc. of concentrated nitric acid. This treatment oxidizes the sodium phosphide to sodium phosphate. The presence of the latter is tested for by adding to the cooled solution twice its volume of ammonium molybdate reagent. The tube is heated to such a temperature that it can just be held in the hand, and is then set aside. The formation of a yellow precipitate indicates phosphorus, provided arsenic is not present. [1] Identification of Pure Organic Compounds, Vol. I. |
|||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||